
Abstract
Fabrication of high-quality semiconductor thin films has long been a subject of keen interest in the photocatalytic field. Here, we report a facile, solution-based anodic plating and calcination for large-scale synthesis of BiVO4 thin films on indium tin oxide coated glass for use as photoanodes in solar water splitting. Using Na2SO3 as a sacrificial reagent, continuous solar H2 production with 94% Faradaic efficiency was obtained over 6 h of photoelectrochemical water splitting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Solar-driven photoelectrochemical (PEC) water splitting is a promising route for the large-scale production of renewable hydrogen fuel from water [1,2,3,4,5]. In the past decades, much effort has been made to improve the overall energy efficiency of PEC devices [6,7,8,9]. In terms of the photocathodes, high photocurrent densities with low overpotentials have been realized using p-type solar-cell materials in combination with hydrogen-evolution co-catalysts. However, the improvement in photoanodes remains limited [10].
Among a range of photoanodic materials, BiVO4 has attracted research attention because it has a deep valance band position for the oxygen evolution reaction [11]. Also, BiVO4 is relatively stable in neutral aqueous environments (pH 7–9) [12, 13]. Over 100-h PEC water oxidation has been reported using crystalline BiVO4 photoanodes [14]. One of the remaining challenges for BiVO4 is to increase the photocurrent density under photocatalytic conditions without applying any external electrical potential. To this end, Choi et al. reported the synthesis of nanoporous BiVO4 photoanodes in a two-step process using BiOI nanoplates as the precursor [15]. Nanostructure certainly improves the charge separation; however, it also presents a difficulty for fabricating a p-n junction that is able to cover the entire BiVO4 to make stand-alone, photocatalytic water-splitting catalysts.
In this work, we report a facile synthesis of BiVO4 thin films on transparent, conductive indium tin oxide (ITO) substrates using anodic plating and thermal calcination. A homogeneous mixture of the anodically deposited bismutite hydrate ((BiO)4(OH)2CO3) and vanadium ions (Fig. S1 in the supporting information) allows nucleation of stichometrical BiVO4 during calcination. Also, bismutite hydrate decomposes at temperatures > 500 °C and releases CO2; the synthesized BiVO4 is thus free of contamination. Using Na2SO3 as a sacrificial reagent, stable photoelectrochemical H2 generation was realized over 6 h of water splitting. The present study shows a promising solution-based process for the preparation of BiVO4 thin films for use in water-splitting applications.
Results and discussion
Layer-structured bismutite and its hydrate were first reported in 1943 [16] and systematically studied in 1984 [17]. In mineralogy, bismutite is a well-established solid carbonate in the system Bi2O3-CO2-H2O [16] with a natural color of yellow to brown. In the laboratory, the synthesis of bismutite has only been reported using the hydrothermal method and with the products in the form of particles [17, 18]. In the present study, we found that anodic plating can also synthesize amorphous Bi4O4(OH)2CO3 thin films on ITO glass via Kolbe electrolysis with the presence of Bi ions, following the Eq. 1:
Figure 1 shows the anodic plating of amorphous Bi4O4(OH)2CO3 films on ITO substrates using NaCOOH and Bi(NO3)3 solutions at pH ~ 5. The applied potential is + 2.3 V vs Ag/AgCl (VAg/AgCl). After 7-min anodic plating, ~ 300-nm-thick film was plated. Likely, the NaCOOH was oxidized at anodic potentials (Eq. 1) to form CO2 on the electrode surfaces. With the presence of Bi3+ in the solution, Bi4O4(OH)2CO3 precipitated on the electrodes. We also found that Bi metals precipitated on the cathode. This is because the reduction potential of Bi3+ to Bi was + 0.2 VRHE, which is more positive than 0 VRHE of the hydrogen evolution reaction. To suppress the Bi precipitation, p-benzoquinone can be added to the electrolyte. The cathodic reaction then mainly shifts to the reduction of p-benzoquinone to 1,4-hydroquinone with a reduction potential of ~ + 0.6 VRHE (Fig. S2). The optical images of the anodically plated amorphous Bi4O4(OH)2CO3 films are provided on the right panel in Fig. 1, which shows the change of color at the different time of the plating.

Schematic of the anodic plating for depositing (BiO)4(OH)2CO3 films on ITO glasses
To understand anodic plating details, Bi4O4(OH)2CO3 films were characterized by scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), UV–Vis diffuse-reflectance analyses and thermogravimetric analysis (TGA) (Figs. 2, 3 and Figs. S3, S4 in the supporting information). To evaluate constituent compositions, the plated Bi4O4(OH)2CO3 films were scratched from ITO/glasses for TGA with a temperature rise from 30 to 500 °C in an N2 atmosphere (N2 was used to avoid adsorption of CO2 from the air). As shown in Fig. 2a, three steps of endothermic decomposition were obtained in a TGA run for Bi4O4(OH)2CO3. A continuous weight loss of 5.6% below 180 °C was observed, likely attributed to absorbed solvent and water [17]. Bi4O4(OH)2CO3 decomposition often occurs in two stages [17]: major loss of hydrate with a small loss of carbon dioxide at 180–240 °C and major loss of carbon dioxide at 240–500 °C. The calculation of weight losses in each step yielded an empirical formulation of (BiO)4(OH)1.01(CO3)0.94 of the anodically plated films, which agreed with the predicted products of Bi4O4(OH)2CO3. As a reference, we also analyzed Bi2O2CO3 powder (Wako, 99.5%) by TGA under the same conditions. Decomposition to release carbon dioxide was observed at 240–500 °C with a weight loss of 9.6 wt%, close to the theoretic value of 8.6 wt%. A slight shift of decomposition onset temperature of Bi2O2CO3 compared to that of plated Bi4O4(OH)2CO3 films was likely due to the crystalline and amorphous nature of the two materials. This result suggested that amorphous Bi4O4(OH)2CO3 films were anodically plated on ITO.

a TG and DTA of a plated (BiO)4(OH)2CO3 film. b TG and DTA of the commercial (BiO)2CO3 (99.5%). c XRD pattern of the as-plated (BiO)4(OH)2CO3, hydrothermal-treated (BiO)4(OH)2CO3, ITO and Bi2O3 calcinated from (BiO)4(OH)2CO3. d UV–vis diffuse reflectance spectra of the as-plated (BiO)4(OH)2CO3, hydrothermal-treated (BiO)4(OH)2CO3, ITO and Bi2O3 calcinated from (BiO)4(OH)2CO3

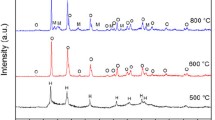
a Optical images of the solution-based process for the synthesis of BiVO4 films on ITO glass (12 pieces of the 2 cm × 2 cm samples). b Schematic diagram of a proposed reaction mechanism. c XRD patterns of the BiVO4/ITO synthesized at 520 °C
Figure 3a shows the optical images of the synthesis process of BiVO4 thin films on ITO substrates. In brief, we deposited amorphous Bi precursors on ITO substrates and calcinated them with the vanadium source at 520 °C. After the reaction, we washed the surface residual vanadium chemicals using 1 M NaOH solution. This process was similar to our previous report, in which the BiVO4 was fabricated via three steps: precursor deposition, pre-calcination of the deposited films in the air at 200 °C, and calcination with a vanadium source at 490–530 °C. We used two-step fabrication, which was able to fabricate BiVO4 thin films with similar morphology. The Bi precursor materials were calcined in the air at 520 °C (Fig. 3c) for 2 h. The fabricated BiVO4 films were in a monoclinic structure, which agreed with the previous report [8].
The scanning electron microscopy (SEM) images of the plated Bi4O4(OH)2CO3 film are present in Fig. S3 in the supporting information. The 7-min anodically plated amorphous Bi4O4(OH)2CO3 film was ~ 300 nm thick on the ITO substrate (Fig. S3). Bi was detected on the surface and in the bulk of the film (Fig. S4). As shown in cross-sectional SEM images in Fig. 4, the BiVO4 film was made of large BiVO4 particles with an in-plane diameter of ~ 500–1000 nm and a thickness of ~ 300 nm. Such large BiVO4 crystalline likely decreased the number of boundaries between particles. Therefore, improved photoelectrochemical performance was realized.

SEM images of BiVO4. a The cross-sectional view of BiVO4. b–d The top view of BiVO4
Finally, we tested the photoelectrochemical performance of the fabricated BiVO4 films in 0.1 M Na2SO3 solution. As shown in Fig. 5a, a quick raise of the photocurrent density was observed with BiVO4 films with the increase of the positive potential. At 0.9–1.2 VRHE, the photocurrent density reached a plateau of ~ 5 mA/cm2 under simulated solar light irradiation. We used a micro gas chromatography (micro-GC) to analyze the hydrogen evolution, which was stable over 6-h photoelectrochemical water splitting at 0.9 VRHE (Fig. 5b).

Photoelectrochemical performance of BiVO4 film in a 0.1 M potassium phosphate (KPi) and 0.1 M Na2SO3 solution. a Photocurrent densities at 0–1.2 VRHE; b evolved H2 in 6 h
Conclusions
In this work, anodic plating was reported for the fabrication of Bi precursors on the indium tin oxide (ITO) substrates. Following high-temperature calcination with vanadium sources, crystalline BiVO4 was fabricated on ITO substrates. The photocurrent density reached ~ 4–5 mA/cm2 at 0.9–1.2 VRHE in Na2SO3-containing electrolytes under simulated solar illumination. The developed electrochemical deposition and thermal calcination may offer a new pathway for the synthesis of photocatalytic materials.
Experimental
Anodic-plating of Bi4O4(OH)2CO3 film on ITO glass
A 0.1 M Bi(NO3)3 solution was prepared by dissolving Bi(NO3)3·5H2O in 25 ml acetic acid solution (99.7% mass ratio, Wako). The prepared solution was mixed with a 10 mL pH 5 sodium acetate aqueous solution. 5 M NaOH solution was used to adjust the pH of the mixed solution to 4.8. Anodic plating was performed at 2.3 VAg/AgCl at room temperature using a three-electrode cell with ITO working electrodes, a platinum counter electrode and an Ag/AgCl reference electrode. A potentiostat (CHI630e) was used for anodic plating and subsequent electrochemical measurements. The optimum plating time was 7 min.
Fabrication of BiVO4 thin film on ITO glass
First, ~ 300 nm amorphous Bi4O4(OH)2CO3 film was anodically plated on an ITO glass in a bismuth nitrite and sodium acetate aqueous solution. Drop-casting vanadyl acetylacetonate (VO(acac)2) organic solutions onto the plated Bi4O4(OH)2CO3 films allowed homogeneous mix of the V and Bi species. The mixed samples were annealed at different temperatures. Finally, the obtained film samples were washed with NaOH to remove impurities. In detail, 0.075 mL 0.2 M VO(acac)2 dimethyl sulfoxide solution was dropped on the Bi4O4(OH)2CO3 films (2 cm × 2 cm) and then calcined in a furnace at 520 °C for 2 h in air. Remained VOx on top of the BiVO4 films was washed in 1 M NaOH solution for 10 min with gentle stirring.
Measurements
The scanning electron microscopic (SEM) images were obtained by a Hitachi SU8020. The XRD diffraction spectra were performed using the smart lab XRD of Rigaku, Japan. The XPS analyses were performed using Mg Kα (1253.6 eV) photon energy. During XPS depth profile studies, Ar bombardment with an etching speed of several tens nm/time was used. Binding energy peak shifts due to any charging were normalized with C 1s peak set to 284.8 eV and Fermi energy position. The TGA analyses were conducted with a differential thermogravimetric analyzer (Rigaku, Japan). The PEC performances were measured by a three-electrode electrochemical configuration with a 0.1 M Na2SO3 solution under simulated sunlight illumination (SAN-EI electronic, XES40S1, AM 1.5G, 100 mW cm−2). An Ag/AgCl electrode was used as a reference electrode, and a Pt coil was used as a counter electrode. The measured potentials were all converted to the reversible hydrogen electrode according to the Nernst equation:
The PEC cell was connected to a vacuum pump and a micro-GC (Agilent 990 micro). Before measurement, the PEC cell was pumped to a low vacuum, and Ar gas was used to purge out the N2 and O2 gases in the cell. The H2 evolution was measured in under simulated sunlight illumination for 6 h at 0.9 VRHE. The theoretical amounts of evolved H2 were estimated from the passed charges on the assumption that faradaic efficiency was unity.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ITO:
-
Indium tin oxide
- PEC:
-
Solar-driven photoelectrochemical
- SEM:
-
Scanning electron microscopy
- XPS:
-
X-ray photoelectron spectroscopy
- XRD:
-
X-ray diffraction
- TGA:
-
Thermogravimetric analysis
References
Grätzel M. Photoelectrochemical cells. Nature. 2001;414:338–44. https://doi.org/10.1038/35104607.
Reece SY, Hamel JA, Sung K, et al. Wireless solar water splitting using silicon-based semiconductors and earth-abundant catalysts. Science. 2011;334:645–8. https://doi.org/10.1126/science.1209816.
Wang Q, Hisatomi T, Ma SSK, et al. Core/shell structured La- and Rh-codoped SrTiO3 as a hydrogen evolution photocatalyst in Z-scheme overall water splitting under visible light irradiation. Chem Mater. 2014;26:4144–50. https://doi.org/10.1021/cm5011983.
Zhong M, Tran K, Min Y, et al. Accelerated discovery of CO 2 electrocatalysts using active machine learning. Nature. 2020;581:178–83. https://doi.org/10.1038/s41586-020-2242-8.
Li L, Ozden A, Guo S, et al. Stable, active CO2 reduction to formate via redox-modulated stabilization of active sites. Nat Commun. 2021;12:5223. https://doi.org/10.1038/s41467-021-25573-9.
** L, Zhao H, Wang ZM, Rosei F. Quantum dots-based photoelectrochemical hydrogen evolution from water splitting. Adv Energy Mater. 2021;11:2003233. https://doi.org/10.1002/aenm.202003233.
Jiang C, Moniz SJA, Wang A, et al. Photoelectrochemical devices for solar water splitting—materials and challenges. Chem Soc Rev. 2017;46:4645–60. https://doi.org/10.1039/C6CS00306K.
Zhong M, Hisatomi T, Minegishi T, et al. Bulky crystalline BiVO4 thin films for efficient solar water splitting. J Mater Chem A. 2016;4:9858–64. https://doi.org/10.1039/C6TA03072F.
Zhong M, Hisatomi T, Kuang Y, et al. Surface modification of CoOx loaded BiVO4 photoanodes with ultrathin p-Type NiO layers for improved solar water oxidation. J Am Chem Soc. 2015;137:5053–60. https://doi.org/10.1021/jacs.5b00256.
Sivula K, van de Krol R. Semiconducting materials for photoelectrochemical energy conversion. Nat Rev Mater. 2016;1:1–16. https://doi.org/10.1038/natrevmats.2015.10.
Andrei V, Jagt RA, Rahaman M, et al. Long-term solar water and CO2 splitting with photoelectrochemical BiOI–BiVO4 tandems. Nat Mater. 2022;21:864–8. https://doi.org/10.1038/s41563-022-01262-w.
Abdi FF, Han L, Smets AHM, et al. Efficient solar water splitting by enhanced charge separation in a bismuth vanadate-silicon tandem photoelectrode. Nat Commun. 2013;4:2195. https://doi.org/10.1038/ncomms3195.
Seabold JA, Choi K-S. Efficient and stable photo-oxidation of water by a bismuth vanadate photoanode coupled with an iron oxyhydroxide oxygen evolution catalyst. J Am Chem Soc. 2012;134:2186–92. https://doi.org/10.1021/ja209001d.
Kuang Y, Yamada T, Domen K. Surface and interface engineering for photoelectrochemical water oxidation. Joule. 2017;1:290–305. https://doi.org/10.1016/j.joule.2017.08.004.
Kim TW, Choi K-S. Nanoporous BiVO4 photoanodes with dual-layer oxygen evolution catalysts for solar water splitting. Science. 2014;343:990–4. https://doi.org/10.1126/science.1246913.
Frondel C. Mineralogy of the calcium phosphates in insular phosphate rock. Am Miner. 1943;28:215–32.
Taylor P, Sunder S, Lopata VJ. Structure, spectra, and stability of solid bismuth carbonates. Can J Chem. 1984;62:2863–73. https://doi.org/10.1139/v84-484.
Dong F, Zheng A, Sun Y, et al. One-pot template-free synthesis, growth mechanism and enhanced photocatalytic activity of monodisperse (BiO)2CO3 hierarchical hollow microspheres self-assembled with single-crystalline nanosheets. CrystEngComm. 2012;14:3534–44. https://doi.org/10.1039/C2CE06677G.
Acknowledgements
The authors acknowledge Prof. Kazunari Domen and his group for assistance in the material fabrication and characterization.
Funding
This work was supported by the National Key Research and Development Program of the Ministry of Science and Technology of China (No. 2020YFA0406102), the National Natural Science Foundation of China (Grant Number 22272078, 91963121), and the Frontiers Science Center for Critical Earth Material Cycling of Nan**g University, “Innovation & Enterpreneurship Talents” plan of Jiangsu Province.
Author information
Authors and Affiliations
Contributions
M.Z. supervised the project. M.Z. and H.J. conceived the idea and designed the experiments. H.J. performed the synthesis, characterizations, and performance tests. M.Z., H.J. and Y.X. wrote the manuscript. All authors discussed the results and assisted during manuscript preparation.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, H., **ao, Y. & Zhong, M. Scalable synthesis of BiVO4 thin films via anodic plating and thermal calcination. Discover Nano 18, 6 (2023). https://doi.org/10.1186/s11671-023-03774-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11671-023-03774-z